fairRNAseq
Title: FAIR science Project’s fair RNA-seq
Author: Shradha Mukherjee
Location: Planet Earth
Nicknames: Shredder, a supervillain for villains and Goldy, a superhero for heroes.
Copyright © 2023 onwards fairwissenschaft and Shradha Mukherjee. All rights reserved.
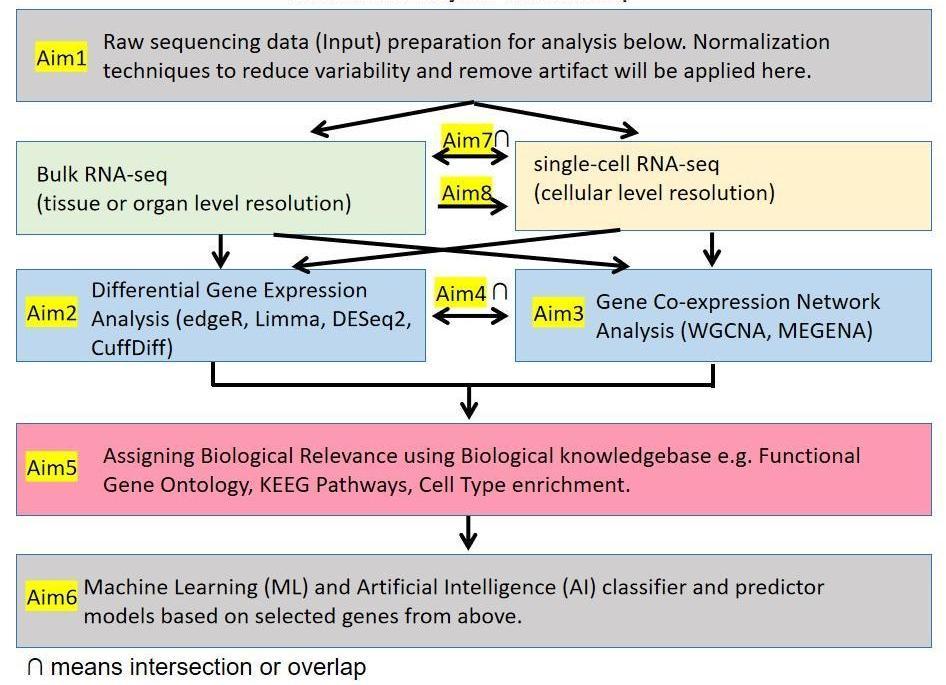
Goal (What?): To develop reproducible integrated bioinformatics pipeline for RNA-seq.
Significance (Why?): For FAIR RNAseq and fair RNAseq.
FAIR is acronym for Findable, Accessible, Interoperable and Reusable. FAIR https://www.ebi.ac.uk/about/news/perspectives/fairification-framework-making-your-data-findable-accessible-interoperable-and-reusable Tier one journals covering all scientific domains and with more than journal published under their umbrella, are Nature, Cell and Frontiers family of journals. Commonly, even when extensive computation is used in their papers, authors do not provide codes during review process and even for final publication Example https://biologicalproceduresonline.biomedcentral.com/articles/10.1186/s12575-022-00175-x. In instances where codes are provided, commonly they are error prone or incomplete, the authors often try to excuse themselves by saying its been a long time since the codes were run, when in reality its not been that long and/or it never actually worked to begin with, Example https://github.com/mgandal/Shared-molecular-neuropathology-across-major-psychiatric-disorders-parallels-polygenic-overlap. Also authors try to excuse themselves from their duty to provide code and data, by stating its human data when in reality de-identified data sharing is standard practice in science. This is a breach of contract with funding agencies who fund the research, who require that once the funded research is completed its published in its entirety (data, methods and results). Works that cannot be verified or reused with citation, are useless for academic, industrial or biomedical science because only rigorous research or FAIR research can be used for applications in human health and environmental health. In biological sciences, say for western blot there is a standard protocol that is used by everyone and any changes needed such as incubation with primary antibody overnight or 6 hours is reported, which makes works that follow this method, verifiable and reproducible, ergo FAIR and useful. Similarly, there needs to be a standard protocol for bioinformatics analysis and this project is an attempt towards that goal. This reproducibility and reusability problem, is not limited to scientific data, techniques and discoveries but also applies to humanities i.e. economics/law/journalism/arts, knowledge domains. Therefore, broadly speaking its not FAIR science peoject but FAIR wissenschaft project.
Fair or just, is giving credit where its due to original work and protecting against plagiarism (use of original work without citation and/or permission). One way to protect against plagiarism is to increase visibility of the original work and make the original work standard practice. Making original work standard practice in science, protects original against plagiarism as original gets applied more in turn gets cited more than plagiarised versions. Example: WGCNA original pipeline https://cran.r-project.org/web/packages/WGCNA/index.html has been extensively plagiarised by other pseudo-WGCNA pipelines that claim to be a new WGCNA invention and repackage a plagiarised version of WGCNA as their own R/bioconductor package. Original WGCNA being a standard well established method has been extensively used to make it robust. These pseudo-WGCNA pipelines create confusion in science, as these pseudo-WGCNA pipelines are error prone and unreliable. Examples of pseudo-WGCNA are, multiWGCNA https://github.com/fogellab/multiWGCNA, GWENA https://bioconductor.org/packages/release/bioc/html/GWENA.html, BioNero https://bioconductor.org/packages/release/bioc/html/BioNERO.html and hdWGCNA https://smorabit.github.io/hdWGCNA, to name a few. If these pseudo-WGCNA represent plagiarism, then what is reasonable use with citation? Sadly, pseudo-WGCNA is more common in science and reasonable use with citation is rare. Infact, I can only give my own example for reasonable use of WGCNA with citation to original WGCNA work. I myself have used WGCNA (https://github.com/smukher2/GithubScientificReportsGlioblastomaStemApril2020, https://github.com/smukher2/GithubSubmittedADPDHDMSimmuneSept2021 and https://github.com/smukher2/GithubFrontiersNeurosciDec2018), and I will use it here in this project, but I cited original WGCNA scientists prominently and never claimed in my publications/presentations/verbally that I invented WGCNA pipeline, novelty of my work being integration of WGCNA into another pipeline and application of this for my research question. This plagiarism problem, is not limited to scientific data, techniques and discoveries but also applies to humanities i.e. economics/law/journalism/arts, knowledge domains. Therefore, broadly speaking its not fair science project but fair wissenschaft project.
This project of developing a standardized RNA-seq pipeline is an effort to make scientific research FAIR (reproducible) and fair (give credit where its due to originals). Funding agencies and journals can ofcourse enforce publication of codes, usage of FAIR science and stop plagiarism, but I am a nobody so I have no control over their decisions/free will.
Specific Aims (What?) and Methods Overview (How?):
Aim1: A) For bulk RNA-seq B) For single-cell RNA-seq (scRNA-seq): To develop an integrated pipeline for converting raw data (sequencer data) to normalized gene expression data.
Aim2: A) For bulk RNA-seq B) For single-cell RNA-seq (scRNA-seq): To develop an integrated pipeline for DGE (differential gene expression) analysis using multiple methods (edgeR, limma, DESeq2, simple comparison of means, cuffDiff).
Aim3: A) For bulk RNA-seq B) For single-cell RNA-seq (scRNA-seq): To develop an integrated pipeline for GCN (gene co-expression network) analysis using multiple methods (WGCNA, MEGENA).
Aim4: A) For bulk RNA-seq B) For single-cell RNA-seq (scRNA-seq): To develop an integrated pipeline to overlap DGE (Aim2) and GNA (Aim3).
Aim5: A) For bulk RNA-seq B) For single-cell RNA-seq (scRNA-seq): To develop an integrate pipeline for interpretation of biological relevance of a gene list i.e. list of genes selected in Aim2, Aim3 and/or Aim4.
Aim6: A) For bulk RNA-seq B) For single-cell RNA-seq (scRNA-seq): To develop an integrate pipeline for Machine Learning (ML) and Artificial Intelligence (AI) classifier models based on biomarker genes i.e. list of genes selected in Aim2, Aim3, Aim4 and/or Aim5.
Aim7: A) From single-cell RNA-seq to bulk RNA-seq: To develop an integrated pipeline for overlapping scRNA-seq and bulk RNA-seq.
Aim8: B) From bulk RNA-seq to single-cell RNA-seq (scRNA-seq): To develop an integrated pipeline for predicting scRNA-seq response based on bulk RNA-seq response using Machine Learning (ML) and Artificial Intelligence (AI) for gene perturbation (e.g. gene editing with Crispr-Cas9), and chemical treatment (e.g. pharmaceutical drugs, environmental toxins).